作者:萧智
“我”想知道粘多糖病它是什么?
“我”是怎么患上粘多糖病的?
“我”并丑陋,我是粘多糖病吗?
“我”的寿命真的就到此为止了嘛?
为什么给“我”这么短暂而又短暂的人生?
难道“我”真的就没救了吗?
——没在这世界待太久的“我”
“我”是一位患有粘多糖病,在这世界上只待了13多月的小男孩。
那天夜班,孩子被抱进来的时候生命体征已经没了,直接死因是呛咳后窒息。这么小的孩子确实很容易呛咳但不是容易窒息而亡的,仔细询问后,原来孩子本身患有粘多糖病。
说实话,对于一个初入临床的“菜鸟”的我来说,真心不知道这病到底是什么,只是看着这小小的身躯,甜甜的脸庞,确实觉得特别的惋惜。内心被触动,所以我要了解这个病,也想要大家了解它。
粘多糖病到底是什么病?
粘多糖病(MPS),也称粘多糖沉积症,其主要是因降解粘多糖(糖氨聚糖GAG)所需要的溶酶体水解酶的缺陷,导致组织内有大量的粘多糖蓄积,从而造成骨骼发育畸形、肝脾肿大、智力障碍和尿中粘多糖排出增多等症状。MPS是一组先天性遗传疾病,各类型粘多糖病的总发病率约为1:250000,MPS患者中男性多于女性,多发生于近亲结婚者的后代,多有家族史。目前,临床上将粘多糖病分为8种类型;其中MPS-Ⅰ型包含3个亚型,后来证实MPS-V型为MPS-IS型,在所有各型粘多糖病中以Ⅰ、Ⅳ型最常见,除Ⅱ型为X性连锁隐性遗传外,其他均属于常染色体隐性遗传病。(在此要细细的了解一下“X性染色体隐性异常”和“常染色体隐性遗传”,见下图。)
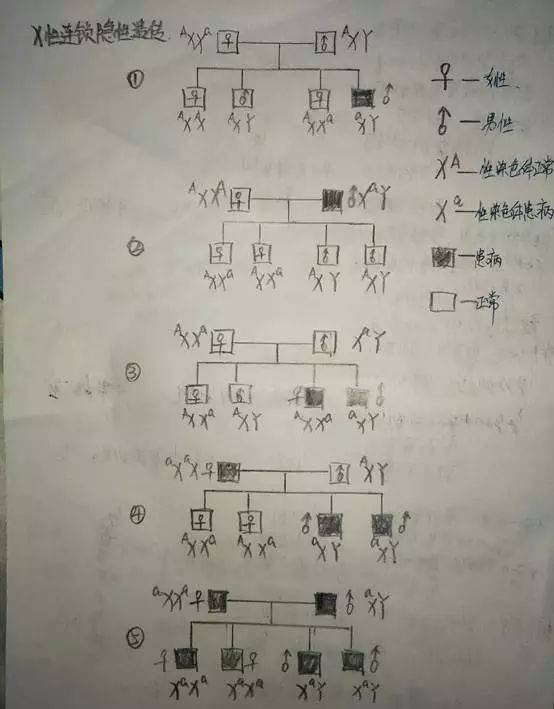
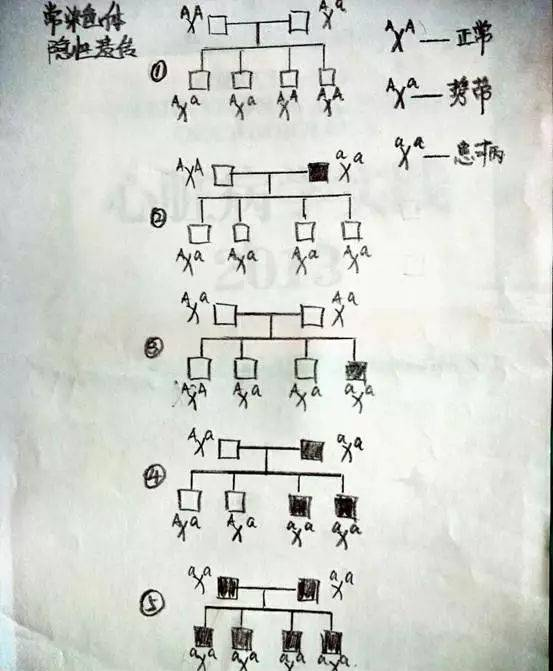
这也就解释了为什么MPS的发病率男性多于女性了。
为什么会患上粘多糖病?
MPS是由于编码各种粘多糖代谢酶的基因发生点突变、无义突变、错义突变、缺失、插入、重复等变异,导致体内粘多糖降解所需的各种酶缺陷,使粘多糖降解代谢发生障碍,从而导致不能降解的各种粘多糖成分蓄积于体内。主要的粘多糖有5种,包括硫酸皮肤素(DS)、硫酸类肝素(HS)、硫酸角质素(KS)、硫酸软骨素(CS)和透明质酸等,它们是人体组织中角膜、软骨、骨骼、皮肤、筋膜、心瓣膜和血管结缔组织的结构成分。过多的粘多糖沉积于上述组织,可引起相应的器官损害及功能障碍,且过多的粘多糖还可从尿液中不断排出。
粘多糖病有多少分型?
各型粘多糖病的酶缺乏及增多的粘多糖成分

因粘多糖病Ⅰ(H)型患者面容丑陋,长相似中国古建筑屋檐下天沟(承霤(liu))上的怪物,因此人们也把此病称为承霤病。

承霤图片(来自网络)
粘多糖病具体为表现什么?
Ⅰ型:可分为MPS I-H (Hurler综合征)、MPS I-S(Scheie综合症).MPS I -HS(Hurler-Scheie综合征)3个亚型。患者一般出生时表现正常,6个月至1岁开始逐渐出生长缓慢,智力低下。头大且前额突出,呈舟状,眼距增宽。角膜混浊较常见,严重者可失明。短颈,耸肩,心瓣膜受累可引起心脏增大与心功能不全。四肢短小,脊柱后凸。关节呈屈曲强直状,活动受限,膝关节、踝关节外翻及足扁平。掌指粗短,可出现腕管综合征。Hurler常有综合10岁前死亡,Scheie综合征及Hurler- Scheie综合征可存活至成年。
Ⅱ型:患者几乎均为男性,女性患者罕见,多于2-6岁起病,根据病情轻重分为A, B两个亚型。临床表现与Hurler综合征相似,但出现时间较晚,病情进展较缓慢。身材矮小及智力低下较Hurler综合征轻。常有进行性听力损害,以心脏受累为主要表现,发展为心脏瓣膜病、冠心病、充血性心力衰竭,多数还伴有阻塞性呼吸暂停综合征、肝脾大等,常于15岁前死亡。B型症状较轻,可无听力损害及骨骼畸形。
Ⅲ型:本型虽然有四种不同缺陷酶,但临床表现相似,主要表现为进行性智力减退。一般4-5岁前智力正常,严重者2-3岁可有智力低下、毛发增多,有听力损害,但无角膜混浊,无腹外病,一般不累及心脏。
Ⅳ型:以身材矮小为突出表现,面容及智力正常,学步及出牙时间较晚,角膜混浊出现较早,听力进行性损害。可有骨骼畸形如鸡胸、驼背、膝外翻、关节挛缩等,伴关节松弛,无关节强直。一般可存活至20-30岁。
Ⅵ型:本型极为罕见,临床表现与I型相似,但患者智力一般正常。一般2-3岁开始出现生长发育迟缓,颅缝闭合较早,可出现脑积水引起的颅高压症状及痉挛性偏瘫。角膜混浊出现较早,听力进行性下降,常有心脏瓣膜病变及腹外病,多数患者存活不超过10岁。
Ⅶ型:此型也极为罕见,以特殊面容和肝脾大为主要特征。特殊面容出生后即可出现,但智力正常,角膜混浊及听力损害较常见,鸡胸、膝外翻等骨骼畸形。多有肝脾大,骨骼发育不良。
Ⅷ型:目前只有个例报告,在表型上,骨骼改变与MPSⅣ型相似。可表现为脊椎扁平、舟状头、漏斗胸。
Ⅸ型:1999年报告了1例由于透明质酸酶基因发生突变引起的溶酶体粘多糖贮积症病例,分类为MPSIX型。此型患者临床表现很轻,仅有轻度的身材矮小和关节周围有软组织肿块,无神经系统及内脏受累表现。
粘多糖病影像学检查可多系统异常表现
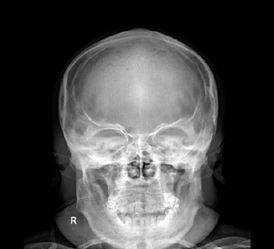
图1头颅顶骨增厚,下领骨增粗增大
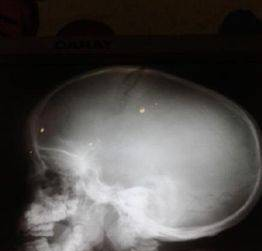
图2蝶鞍“J”形增大
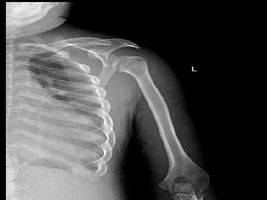
图3肋骨呈“飘带状”,胧骨端肥大肩关节间隙增宽,胧骨头向下移位

图4椎体变扁,前缘变尖呈“鸟嘴样”,椎间隙增宽,椎弓根较细,脊柱后突
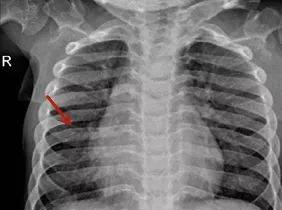
图5肋骨呈“飘带状”
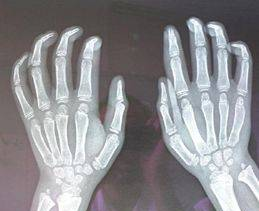
图6骨化延迟,骨化中心仅7枚,双手远端关节呈爪状
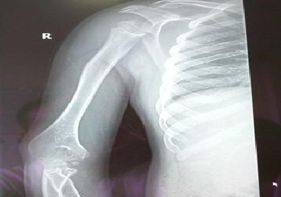
图7胧骨干角稍增大,呈胧外翻

图8尺挠骨远端相对关节面斜形改变

图9左侧股骨远端、胫骨近端骨端肥大
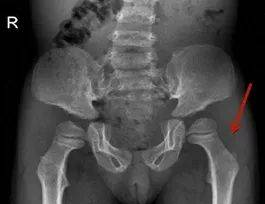
图10骼骨翼、骸基底部变尖,骸臼变浅股骨头扁小,双骸外翻畸形
粘多糖病如何诊断?
1.典型的临床表现:严重的智力障碍、特殊丑陋面容、肝脾及心血管等多器官受累、骨骼畸形等;
2.实验室检查:尿粘多糖定性实验阳性、24小时尿粘多糖>100mg/24h,酶活性的测定;
3.骨骼影像学改变:如头颅增大,脊柱后凸畸形,椎体呈“子弹头”或“鸟嘴”样改变,飘带状肋骨,四肢骨骼畸形,关节外翻以及骨化延迟等表现;但病因诊断赖于酶检测及基因分析。
粘多糖病与哪些疾病容易混淆?
1.粘脂病
属于常染色体隐性遗传的溶酶体病。由于溶酶体内多种水解酶缺陷致使粘多糖与糖脂类在体内累积致病,临床与X线与MPS有很多相似之处,尿中可有涎酸单糖排泄量增加,但尿粘多糖阴性。
2.佝偻病
由于骨支撑能力减弱,长管骨常有弯曲畸形,呈膝内翻或膝外翻,胸廓呈鸡胸,肋骨骨质稀疏,前端与软骨交界处膨大如串珠,颅骨因门延迟闭合,头呈方形。粘多糖Ⅳ型如具备鸡胸、X型腿、侏儒,易误诊为佝偻病,临床医师看到鸡胸切勿只想到佝偻病,应注意询问患儿喂养史,实验室检查应注意查血钙磷、尿粘多糖电泳分析以及X线长骨像等可鉴别。
3.软骨发育不全
智力正常,生长发育迟缓,可有鸡胸、骨髓端大,X型腿等,似Ⅳ型粘多糖病,但头型特殊:上半部大且长,躯干长而四肢粗短,X线长骨像与MPS Ⅳ型不同,且掌指骨粗短如哑铃状,椎弓根间距离上宽下窄(粘多糖病人为上窄下宽),尿中粘多糖阴性。
粘多糖病不能治愈但能够治疗
1.对症治疗
1)血浆治疗
输入正常人血浆治疗可使尿中粘多糖排泄减少,血浆中可供给适量的酶。以50mg/kg血浆每4-12周输注一次可改善临床症状,且治疗越早,效果更好。
2)青霉胺治疗
因粘多糖为成纤维细胞所产生,而青霉胺可能使成纤维细胞成熟过程受阻,从而能使粘多糖减少。使用青霉胺治疗后患者的尿中粘多糖排泄均明显减少,但临床疗效尚待继续观察。
2.酶替代治疗(ERT)
酶替代疗法(ERT)用作MPSI型的主要治疗手段。ERT已被证明是一种安全、有效治疗的治疗,能改善患者呼吸功能,关节的灵活性,行走能力,以及生活质量。然而因拉罗尼酶(laronidase,一种抗Ⅰ型粘多糖病的酶)并不穿过血脑屏障,因此在MPSⅠ型中不能防止认知能力下降。长期酶替代治疗对心肌有益,但对心脏瓣膜病变却无明显改善。
3.造血干细胞移植(HSCT)
异基因造血干细胞移植可以替代MPS各型酶的缺陷,能明显改善粘多糖病人病情及延长寿命,是粘多糖病最有效的治疗手段。各类型的MPS因累及的系统及组织存在差异,因此造血干细胞移植疗效也不同。移植前后应注意加强免疫抑制治疗,减低剂量的预处理方案,以及适当增加供者造血干细胞输注数量,有利于促进植入、降低减少移植物衰竭以及移植物抗宿主病发生。早期诊断、早期移植干预治疗是取得良好疗效的关键。
4.基因治疗
因酶替代疗法、造血干细胞移植等都有一定的局限性,因此真正意义上的基因治疗已成为该研究领域的主攻方向。目前,MPS Ⅰ的基因治疗主要是采用基因的修饰和替代疗法,动物实验和临床试验已获得了安全的结果,其临床治疗效果也得到了肯定。真正的基因治疗包括正常基因替代疗法和原位修复IDUA基因,而后者是治疗MPS Ⅰ型患者及携带者的根本手段。
粘多糖病可以预防吗?
本病至今仍无有效的根治疗法,且疗效有限,因此对已有先证者的家庭或有疑似MPS患儿的母亲再次妊娠时可进行产前诊断,为降低该类出生缺陷患儿出生率、减轻社会及家庭负担均有重要的意义。
参考文献:
[1]郭奕斌. 黏多糖病的诊断及防治策略[J]. 2009.
[2].畅智慧, 刘兆玉. 粘多糖病影像学的多系统表现 1 例[J]. 医学影像学杂志, 2010, 20(4): 476-476.
[3].焦丽, 郭新莉, 陈靖, 等. 小儿粘多糖病及粘脂病 21 例误诊分析[J]. 武警医学, 2003, 14(3): 177-178.
1.《你对“粘多糖病”知多少?》援引自互联网,旨在传递更多网络信息知识,仅代表作者本人观点,与本网站无关,侵删请联系页脚下方联系方式。
2.《你对“粘多糖病”知多少?》仅供读者参考,本网站未对该内容进行证实,对其原创性、真实性、完整性、及时性不作任何保证。
3.文章转载时请保留本站内容来源地址,https://www.lu-xu.com/guoji/16558.html