
1.天使综合症
阿吉利斯公司更新天使综合症基因治疗项目

阿吉利斯生物治疗公司最近宣布,技术运营高级副总裁金基宏博士在日本儿科神经病学学会第60届年会上发表了题为“治疗天使综合征的基因治疗方法”的演讲。
金博士发表了阿吉利斯的两位合作者,南佛罗里达大学的埃德温·威伯博士和凯文·纳什博士最近的体内数据。利用该公司的靶向微剂量平台,为基于腺相关病毒的天使综合征(AAV)提供基因治疗angel砷,显示出中枢神经系统的强大生物分布(CNA)。AGIL-阿斯是处于临床前发展阶段的基因治疗研究。金博士还概述了正在进行的制造和非临床活动,这将支持该公司对will阿司特新药的临床研究申请,该申请预计将于明年提交给FDA。
天使综合征是一种罕见的由UBE3A基因缺失或突变引起的遗传病。UBE3A编码泛素连接酶E6-AP,在中枢神经系统功能中起重要作用。安吉尔综合征的特征是发育迟缓、智力迟钝、严重的言语障碍、癫痫和共济失调,可导致慢性残疾,需要终身护理。天使综合症治疗基金会估计,每15000名新生儿中就有一名患有这种疾病。
2.法布里
阿米库斯公司开始在日本销售一种法布里病药物——加拉斯塔德
Amicus Therapeutics最近宣布,已开始将口服Galafold胶囊123 mg (migalastat)商业化,该胶囊用于治疗16岁及以上具有顺应性突变的Fabry病(α-半乳糖苷酶A缺乏症)患者。加拉福是日本第一种也是唯一一种治疗法布里病的口服精确药物。
法布里病(Fabry's disease)是一种潜在致命的罕见遗传性疾病,由于α-半乳糖苷酶A的功能障碍或缺乏而导致溶酶体中疾病底物(酰基鞘氨醇三己基糖,GL-3)的积累,Galafold通过稳定机体功能障碍的酶起作用,从而清除患者体内积累的疾病底物。依从性突变是基于一种对米格拉stat治疗有反应的专有体外试验(Galafold依从性试验)。
3.兰伯特-伊顿肌无力综合征
美国食品和药物管理局接受了NDA用催化剂公司的弗达普斯治疗兰伯特-伊顿肌无力综合征,并给予优先检查权
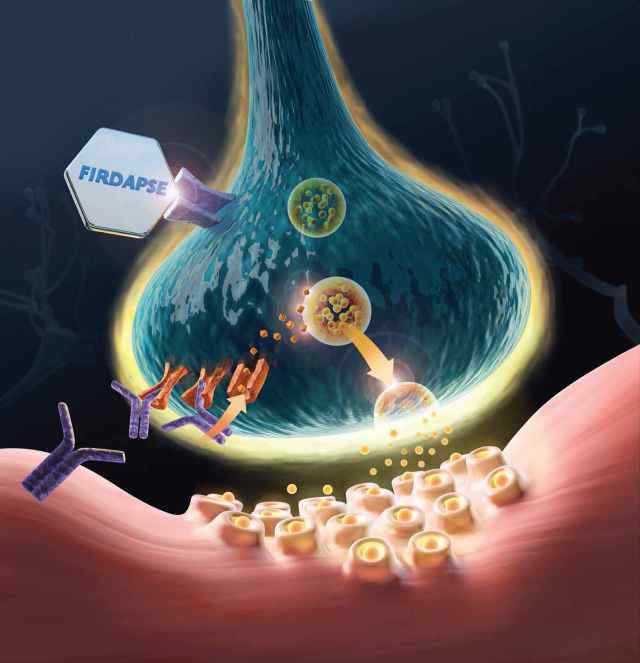
催化剂制药公司宣布,美国食品和药物管理局已经接受了弗达普斯针对兰伯特-伊顿肌无力综合征(LEMS)的新药上市申请(NDA),并进行了优先审查。
该申请得到两项三期试验阳性结果的支持,FDA将于2018年11月28日决定是否批准该药物。
美国食品和药物管理局已将磷酸氨氯地平授予LEMS的突破性治疗资格,以及LEMS、CMS和重症肌无力的孤儿药资格。Firdapse是第一个也是唯一一个欧洲批准的针对成年LEMS患者的对症治疗。
Catalyst还在开发用于治疗难治性婴儿痉挛的CPP-115。CPP-115被FDA授予治疗婴儿痉挛的孤儿药物资格,也被欧洲委员会授予治疗西氏综合征的孤儿药物资格。此外,Catalyst公司正在开发Sabril(viga Bart in)的通用版本。
4.x连锁低磷血症(XLH)
Crysvita治疗儿童X连锁低磷血症优于口服磷酸盐和活性维生素D

Ultragenyx制药公司宣布Crysvita (burosumab)的3期研究已经达到主要终点。经过40周的治疗,Crysvita在改善X连锁低磷血症儿童佝偻病方面优于口服磷酸盐和活性维生素D(常规治疗)(LS平均治疗差异为1.14,P
研究还表明,Crysvita治疗可以改善重要的代谢和功能指标,其安全性与其他Crysvita儿科XLH研究相似。Crysvita是一种阻断成纤维细胞生长因子23(FGF23)的抗体。FGF23是一种能引起磷酸尿排泄,抑制肾脏产生活性维生素d的激素。
这项研究达到了它的主要终点。三名独立的儿科放射科医生采用RGI C量表进行盲目评估。与常规治疗相比,Crysvita能显著改善佝偻病的症状。此外,72%接受Crysyita治疗的患者观察到显著的恢复(RGI-C≥2.0),而接受常规治疗的患者只有6%。
2018年4月17日,美国食品和药物管理局批准Crysvita用于治疗一岁以上的成年人和XLH儿童患者。2018年2月23日,欧洲委员会授予Crysvita有条件营销授权,治疗骨骼生长一年及以上且影像学证据显示有骨病的XLH儿童和青少年。该3期儿科临床试验将在欧洲用作验证研究,但美国的监管应用程序并不要求这样做。
5.大疱性表皮松解症
纤维细胞公司报告了FCX-007治疗大疱性表皮松解症1/2期临床试验的中期结果

纤维细胞科学公司最近更新了FCX-007治疗隐性营养不良性大疱性表皮松解症1/2期临床试验的中期结果和进展。来自患者的安全性数据显示,FCX-007在给药52周后耐受性良好。无严重不良事件和产品相关不良事件报告。无ⅶ型胶原自身抗体反应。
FCX-007是RDEB临床阶段基因治疗的候选产品,RDEB是由COL7缺乏引起的先天性和进行性孤儿皮肤病。FCX-007是一种编码COL7基因的转基因自体成纤维细胞,由Fibrocell和Rercicigen(Introxon的全资子公司)开发。
美国食品和药物管理局已授予FCX-007治疗营养不良性大疱性表皮松解症(包括RDEB)的孤儿药资格。此外,美国食品和药物管理局还授予FCX-007治疗罕见儿科疾病的资格和快速康复医疗中心。
6.威廉姆斯-伯伦综合征
通过面部分析技术成功地识别了威廉姆斯-伯伦综合征
图片来源:NHGRI-Williams综合征
根据2018年5月发表在《美国医学遗传学杂志》上的一项国际研究,研究人员已经成功地使用面部分析技术和临床信息来识别不同人群中的威廉姆斯-伯伦综合征。这项研究由国家人类基因组研究所(NHGRI)领导。
威廉姆斯-贝伦综合征的发病率估计约为1/10000-1/7500。症状和体征包括精神发育迟滞和独特的面部特征,包括眼周水肿、短鼻宽鼻、两颊饱满、大嘴厚唇。
使用面部分析技术,研究人员将非洲、亚洲、高加索和南美洲的286名威廉姆斯-伯伦儿童和成人与286名相同年龄、性别和种族的健康人进行了比较。他们能够以95%或更高的准确率正确识别每个种族的患者。
根据2017年9月、2017年4月和2016年12月发表的研究,该技术在识别努南综合征、迪乔治综合征和唐氏综合征方面也非常准确。本系列的下一项研究将重点关注Cornelia de Lange综合征。
7.庞贝氏病
电子治疗师降低了成年庞贝氏病患者轮椅依赖的风险
根据一项正在进行的调查的最新结果,酶替代疗法(ERTs)可以降低成年庞贝病患者对轮椅的依赖风险。
这项调查的结果发表在5月22日的《俄耳甫尼特罕见疾病杂志》上,文章题为:“酶替代疗法降低了成年庞贝氏病患者对轮椅的依赖风险”。这项前瞻性国际研究旨在调查ERT是否能降低患者需要轮椅、呼吸器或两者兼有的风险。"
虽然这些发现不能证明ERT对呼吸支持的作用,但与历史数据一致,都显示了这种治疗方案对运动结果的积极临床益处。然而,有些病人在生活的某些时候仍然依赖轮椅。“因此,尽管ERT在治疗成人庞贝病患者方面显示出积极的临床效果,但我们也得出结论,仍有改进的余地空”
8.莱特综合征和脆性X染色体综合征
纽伦宣布trofinetide在日本获得了第一项专利
纽伦制药宣布,日本专利局授予trofinetide第一项专利。这项名为“使用甘氨酸-L-2-甲基-L-谷氨酸治疗自闭症谱系疾病”的新专利涵盖了自闭症、雷特综合征和脆性X综合征患者。该专利将于2032年1月到期,并可能延长5年。
Trofinetide在美国和欧洲获得了材料成分专利,在美国、欧洲和澳大利亚获得了治疗Leiter综合征、脆性X综合征等自闭症谱系疾病的专利。加拿大、巴西和以色列的申请正在等待批准。
9.雷特综合征
美国食品和药物管理局授予AMO公司治疗莱特综合症的AMO-04孤儿药资格
AMO制药有限公司是一家私人生物制药公司,专门研究缺乏或没有治疗的罕见衰弱性疾病,最近宣布美国食品和药物管理局(FDA)授予AMO-04研究疗法治疗罕见儿童雷特综合征的孤儿药物资格。
莱特综合征是一种神经系统疾病,通常发生在婴儿期,几乎总是发生在女孩身上。大多数病例是由X连锁甲基化-CpG结合蛋白2(MECP2)基因突变引起的。该病常被误诊为自闭症、脑瘫或非特异性发育迟缓,因为患者往往存在认知、感觉、情绪、运动和自主功能问题。其他症状包括癫痫发作、清醒时呼吸模式紊乱、脊柱侧凸和睡眠障碍。
1.《天使人综合症 10余种疾病:天使综合征、法布雷、庞贝病、雷特综合征、EB等相关研究进展 | YAO闻快讯》援引自互联网,旨在传递更多网络信息知识,仅代表作者本人观点,与本网站无关,侵删请联系页脚下方联系方式。
2.《天使人综合症 10余种疾病:天使综合征、法布雷、庞贝病、雷特综合征、EB等相关研究进展 | YAO闻快讯》仅供读者参考,本网站未对该内容进行证实,对其原创性、真实性、完整性、及时性不作任何保证。
3.文章转载时请保留本站内容来源地址,https://www.lu-xu.com/caijing/1012922.html



