三
微RNA/SIRT1/NF-κB
高血糖状态下,MiR-23 b-3p在微血管内皮细胞(mECs)的外层表达。它下调视网膜内皮细胞(RECs)中SIRT1的表达,并在dr下促进NF-kB的乙酰化水平。
此外,高糖增加了NF-κB P65与编码成熟miR-23b-3p的miR-23b-27b-24-1基因启动子区的结合。因此,通过减少SIRT1蛋白,miR-23b-3p的表达被下调,NF-kB的乙酰化水平被抑制,从而控制了dr的炎症和作用。
高糖诱导氧化应激,调节纤连蛋白(FN)等细胞外蛋白(ECM)的增加,是DR的主要特征。
FN的表达水平在老化的视网膜中非常常见。高糖增加了miR-195的表达水平,导致miR-195与SIRT1的3个非翻译区结合,降低了SIRT1的表达,从而调节衰老的微血管通透性和糖尿病中FN的表达水平。
MiR195可以降低SIRT1的表达,引起P300和组蛋白乙酰化的增加。这可能会导致血管内皮生长因子产品的增加。
四
SIRT1/ET-1,TGF-β1
内皮素1(ET1)和转化生长因子1(TGF-β1)是两种血管活性生长因子,与糖尿病视网膜病变的发展有关,由血管内皮细胞分泌,对维持体内血管张力、整合和平衡至关重要。

高血糖增加ET-1和TGF-B1的表达水平,通过改变血流水平和增加血管通透性引起血管损伤。血管纤维的产生可以通过增加细胞外基质的蛋白质水平来诱导。已经发现SIRT1通过SMAD脱乙酰化增加TGF-β1的表达水平并下调ET-1的表达,以防止纤维化的形成。
五
缺口/SIRT1
Notch信号是一种关键的细胞内信号机制,它协调血管的生长。虽然改变Notch信号活性足以引起内皮细胞行为和血管生成的差异,但对内皮细胞Notch信号的研究很少。
NAD+-依赖的SIRT1脱乙酰化可以负调节内皮细胞的Notch信号。激活Notch1的胞内结构域(NICD),抑制Notch 1的脱乙酰化。
六
SIRT1/HIFs
氧在dr中起重要作用,视网膜中的血管内皮生长因子可由缺氧诱导因子(HIFS)调节。缺氧诱导因子是一种转录因子,在调节代谢中起重要作用。
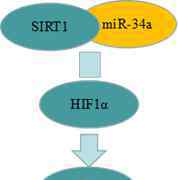
高血糖时稳定视网膜中的缺氧诱导因子-1/2α可增加血管内皮生长因子的表达。MiR-34a的过表达抑制了SIRT1的表达,提高了HIF1α的表达水平,促进了细胞凋亡。
此外,高糖可诱导SIRT1表达水平升高,抑制HIF-1α表达水平。因此,SIRT1调节微血管再生的部分原因是缺氧诱导因子-1α和缺氧诱导因子-2α失活。
七
SIRT1/IL-17
TH-17细胞和白介素-17参与炎症反应,与糖尿病视网膜病变的发展有关。白介素-17在糖尿病视网膜病变组织中的表达增加。白介素-17由TH17分泌,是中性粒细胞活化和迁移的关键细胞因子。
此外,IL-17可以诱导和分泌非免疫细胞分泌促炎因子。通过作用于ECs细胞,打破紧密联系和血脑屏障,促进血管内皮生长因子的表达和血管再生。所以IL-17增加了视网膜损伤。
外周血单个核细胞中IL-17与SIRT1的平衡可能导致dr的病理形成,在DR中,SIRT1被激活后,IL-17的表达水平受到抑制,限制了炎症的进程。
八
PRMT1/SIRT1
氧化应激介导的血视网膜屏障(BRB)损伤与DR的进展有关,视网膜色素上皮细胞易受氧化应激的影响。视网膜毛细血管内皮细胞是BRB内叶的重要组成部分。
PRMT1被认为与dr有关。ROS在这两种细胞系中的表达水平增加。总之,氧化应激被SIRT1下调,诱导PRMT1的表达,增加视网膜色素上皮细胞的凋亡。
九
GLP-1R/SIRT1/P53
在糖尿病患者中,肠道内分泌的GLP-1通过GLP受体调节血糖。GLP-1R是一种G蛋白偶联受体,保护神经元免受凋亡。GLP-1R减轻视网膜氧化应激和防止细胞凋亡和GLP模拟。
高血糖易导致视网膜神经元和穆勒神经元产生一氧化氮。一氧化氮和超氧化物共同产生过氧硝酸盐,导致视网膜损伤和死亡。ROS增加可导致视网膜神经变性和视力障碍。
SIRT1是GLP-1的激动剂,可保护视网膜神经元。GLP-1受体激动剂增加了NAD/NADPH的比例,增加了SIRT1的表达,增加了超氧化物的清除,但抑制了氧化应激反应。
它减少细胞凋亡和炎症因子,保持BRB的完整性。SIRT1介导的抗凋亡作用增加了P53的脱乙酰化。在视网膜色素上皮细胞中,乙酰化p53导致促凋亡BCL-1家族蛋白的激活,包括BAX和PUMA。因此,P53-BAX和P53-PUMA复合物转移到线粒体,激活caspase-9和caspase-3,最终导致细胞凋亡。
10
基质金属蛋白酶-9/全身炎症反应综合征1/核因子κB
糖尿病患者视网膜毛细血管内皮细胞基质金属蛋白酶-9水平升高。在糖尿病中,氧化应激的增加抑制了SIRT1的表达。NF- κB和AP-1的乙酰化增加了它们与MMP-9启动子区的结合。

在线粒体中,活化的MMP-9损伤细胞膜,增加氧化应激,加速视网膜毛细血管的凋亡。SIRT1的过度表达降低了它们的乙酰化和与MMP-9启动子区的结合,最终降低了MMP-9的转录。
11
AMPK/SIRT1/NF-κB
腺苷磷酸活化蛋白激酶(AMPK)是一种保守的丝氨酸苏氨酸蛋白激酶,控制下游因子的表达和活化。AMPK通过增加烟酰胺磷酸核糖激酶(NAMPT)和NAD+的表达来激活SIRT1。SIRT1通过乙酰化和激活LKB1激活AMPK信号通路。
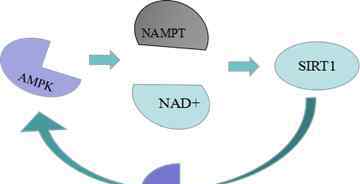
AMPK的激活类似于SIRT1去乙酰化NF-kB,SIRT1在AMPK去乙酰化NF-kB并抑制它的信号通路中非常重要。
此外,AMPK增加了ICAM-1的表达和视网膜血管中的白细胞浸润,AMPK和SIRT1的激活通过NF-kB乙酰化抑制ICAM-1和血管内皮生长因子的转录和表达。

12
SIRT1/FOXO1
SIRT1的缀合物之一是FOXO转录因子。与促进细胞凋亡相比,FOXOs促进细胞存活。
SIRT1促进FOXO基因表达,包括在发生凋亡时降低基因转录。因此,SIRT1似乎与细胞死亡和应激抵抗无关。总之,高糖通过FOXO1降低SIRT1的表达,增加氧化应激。此外,FOXO1可以通过SIRT1激活来预防。
13
AGEs/SIRT1
AGEs遇到高糖时会糖化蛋白质或脂类。AGEs主要集中在糖尿病患者的视网膜血管细胞、神经元和胶质细胞,最终导致DR病变。
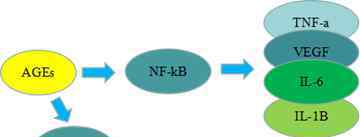
AGEs调节SIRT1蛋白和mRNA水平的下调。能激活NF-κB的转录,促进ICAM-1等黏附分子的表达,增强TNF-a、VEGF、IL-6、IL-1B等细胞因子的分泌。需要进一步研究在AGEs中由SIRT1促进的促炎因子和趋化因子。
刚进实验室的时候,不知道从哪个渠道入手。这13个套路足够你用一年了。
1.《sirt1 SIRT1承包了糖尿病视网膜病变(DR)一年的套路》援引自互联网,旨在传递更多网络信息知识,仅代表作者本人观点,与本网站无关,侵删请联系页脚下方联系方式。
2.《sirt1 SIRT1承包了糖尿病视网膜病变(DR)一年的套路》仅供读者参考,本网站未对该内容进行证实,对其原创性、真实性、完整性、及时性不作任何保证。
3.文章转载时请保留本站内容来源地址,https://www.lu-xu.com/fangchan/657661.html





