高效液相色谱法(HPLC)常用于检查药物的有关物质,其计算方法有外标法、带校正因子的自控法、自控法和峰面积归一化法。其中带校正因子的自控法准确,不需要每次都提供杂质对照品,是目前常用的方法。这里简要介绍校正系数的测量和校正系数准确度的验证。
一、修正系数的确定
校准因子采用单点法、多点法、标准曲线法和吸收系数比值法确定。很多文献都报道了每种方法的基本操作和优缺点,这里不再赘述。在确定校正系数时,边肖建议使用标准曲线法,因为线性试验是方法验证的重要组成部分。使用标准曲线法不仅验证了杂质对照品和主成分的线性,而且获得了准确的校正因子,一举两得。
确定校正因子时,首先将各杂质对照品和主成分对照品配制成一定浓度的原液,逐渐稀释,直至找到各杂质的定量限。线性范围应包括定量限浓度~限浓度的150%,主成分浓度范围应包括定量限浓度~自身对照浓度的150%。例如在有关物质的测定中,供试品浓度为1mg/ml,采用0.1%的自身对照(即自身对照的浓度为1μg/ml),杂质A的限度为0.1%(即杂质A的浓度限度为1μg/ml)。假设主成分和杂质A的定量限均为20ng/ml,那么杂质A和主成分应在20 ng/ml ~以内。绘制出各杂质和主成分的线性曲线后,各杂质和主成分之间的线性方程的斜率之比即为杂质的校正系数。如果杂质校正系数在0.9~1.1范围内,则直接采用自参比法,无需验证。如果杂质的校正系数在0.2~5.0之间,则需要有校正系数的自参方法。如果杂质的校正系数小于0.2或大于5.0,杂质的检测波长可以改变并重新确定。如果所有测量值都在0.2~5.0之外,则表示添加了校正系数的自参考。
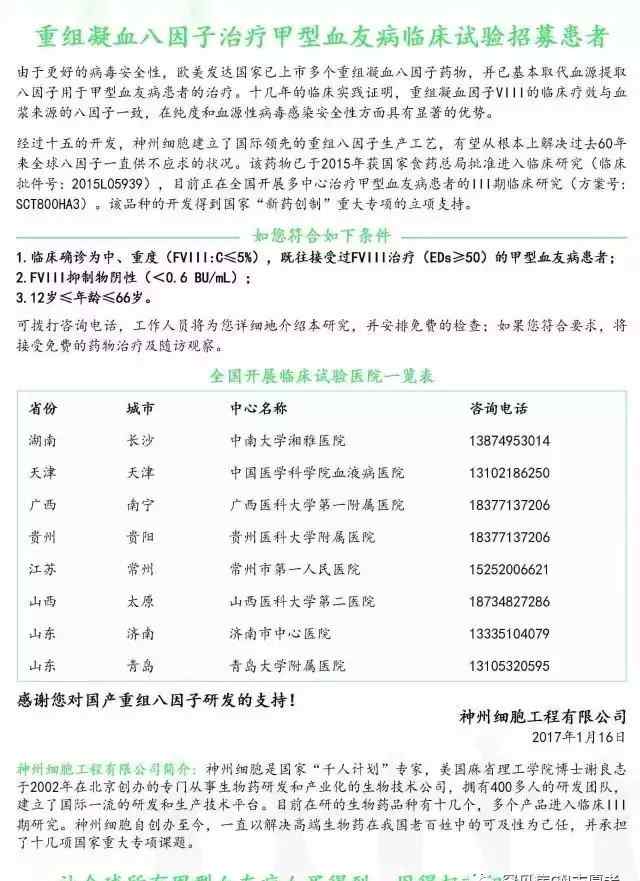
配制各种浓度的线性溶液时,一定要注意需要逐步稀释。为了避免下面图1所示的情况,我们应该在图2中显示它,以确保各个点分布的均匀性。

图1不正确的线性测试设计

图2正确的线性测试设计
第二,相对校正系数和相对保留时间的持久性
当使用带有校正因子的自控方法进行定量时,通常通过相对保留时间来定位杂质。此外,还研究了相对校正因子和相对保留时间的持久性。
具体考察方法与其他方法验证的耐久性试验相同,即改变波长、流速、柱温、流动相比例、流动相pH值、使用不同品牌的色谱柱和仪器等。由各杂质对照品和主成分制备混合对照溶液。用标准曲线法绘制了杂质对照品和主成分对照品在不同条件下的线性方程,并计算了校正因子。同时,计算了不同条件下杂质对照品相对于主成分的相对保留时间。
至于相对校正因子,除了波长的变化,其他色谱条件的调整不会影响相对校正因子。因此,测定波长应选择在最大吸收或最小吸收的位置,而不是吸光度快速变化的位置。关于修正系数变化范围的可接受范围,目前尚无定论。边肖认为,如果最终标准中的相对修正系数只保留到小数点后一位,如0.7,则修正系数在所有情况下都控制在0.65~0.74的范围内,说明耐久性好。也就是说,采用有效数字的修订规则,修订值是标准中规定的值。
对于相对保留时间,除了波长的变化,其他条件的变化都会对其产生影响。为了确保杂质的准确定位,必须研究相对保留时间的持久性。这里的测试不是为了证明相对保留时间的持久性,而是为了确定如何利用相对保留时间来定位杂质。比如固定色谱柱的包装和品牌,固定主峰的保留时间。至于RRT的可接受范围,不能一概而论。如果杂质谱复杂,质量标准中的RRT需要保留到小数点后两位。如果杂质谱比较简单,杂质之间的距离很远,RRT可以保持到小数点后一位。在这里,我想和大家分享另一个经验,就是如果某些杂质的相对保留时间不能固定,并且随色谱参数的变化很明显,可以定向破坏主成分,通过系统适用性试验来定位杂质。
第三,调查相对修正系数的准确性
配制相同的供试品溶液,用自制法和带校正因子的外标法测定供试品中的杂质含量。比较两种方法的测定结果。校正系数与外标法的负偏差不得超过0.05%,正偏差不得规定。如果试样中检测到的杂质很少,可以向试样中加入一定量的杂质对照品进行测定。
此时,可以将相对校正因子固化到质量标准中,用带校正因子的自控方法定量计算已知杂质,既保证了定量结果的准确性,又简化了试验,节省了对照品的消耗。
1.《校正因子 杂质相对校正因子的测定——实战篇》援引自互联网,旨在传递更多网络信息知识,仅代表作者本人观点,与本网站无关,侵删请联系页脚下方联系方式。
2.《校正因子 杂质相对校正因子的测定——实战篇》仅供读者参考,本网站未对该内容进行证实,对其原创性、真实性、完整性、及时性不作任何保证。
3.文章转载时请保留本站内容来源地址,https://www.lu-xu.com/jiaoyu/1618205.html