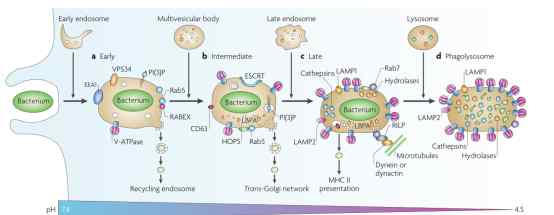
单细胞测序技术于2009年问世。自2013年被《自然方法》杂志评为年度技术以来,它在科学研究中的应用越来越多。
2015年以来,10X基因组学、Drop-seq、Micro-well、Split-seq等技术的出现,彻底降低了单细胞测序的成本门槛。
此后,单细胞测序技术被广泛应用于基础科学研究和临床研究。单细胞在许多领域都占有一席之地,对癌症的早期诊断、随访和个体化治疗具有重要意义。
一、基本原则
首先,单细胞测序并不是只对一个细胞进行测序,而是可以对单个细胞的基因组或转录组进行测序,可以理解为单细胞水平的测序。
在介绍基本原理之前,我们先试着回答一下:为什么要对单细胞进行测序?换句话说,单细胞测序技术能解决哪些传统方法无法解决的问题?
世界上没有两片完全相同的叶子。对于多细胞生物来说,细胞之间是有区别的,往往是基因组和转录组上的几分钱,功能上的几千里。
例如,在肿瘤组织中,肿瘤中心的细胞和肿瘤周围的细胞之间,以及原发灶和转移灶的细胞之间,存在基因组和转录组等遗传信息的差异,从而导致免疫特性、生长速度、侵袭能力等方面的表型差异。并最终导致对不同抗肿瘤药物或放疗敏感性的不同。
那么我们如何研究遗传信息的异质性呢?传统的测序方法是在多细胞水平上进行的,这使得每个人都丢失了异质信息,单细胞测序可以很好的解决这个问题。举个更生动的例子:
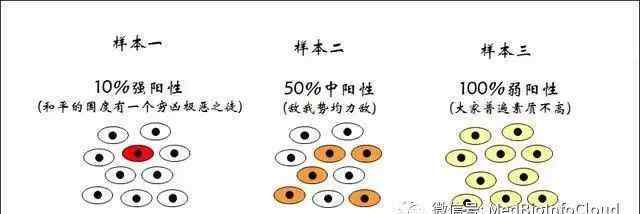
蛋白质印迹检测

虽然这三个样本之间有很多差异,但我们得出的结论是,该基因在不同组织中的表达基本相同。上图所示的异构信息完全忽略。
与western blot类似,传统测序方法显示的信息也是多细胞水平的平均信息,而单细胞水平的测序可以完全反映同一细胞群中不同细胞的基因组和转录组。
随着单细胞测序技术的出现,从混合样本中筛选异质信息的问题得以解决,该技术的成熟应用必将使生命科学研究向前迈出一大步。
那么单细胞测序是如何实现的呢?以单细胞RNA-seq为例,我们简单介绍一下实现该技术的原理:
1.分离单细胞,分别构建测序文库,测序。这种思维通量极低,成本极高。如上所述,烧很多钱会测几十个细胞,往往不足以反映真正的科学问题。所以我们把重点放在第二个选项上。
2.基于标签的单细胞识别。它的核心思想是在逆转录过程中给每个细胞添加一个独特的标签序列,然后对其mRNA进行测序。这样,即使我们混合测序,也可以认为携带相同标签序列的RNA片段来自同一细胞。利用这种策略,我们可以通过一次建立数据库来测量数万个单细胞的信息。

单细胞测序给大家带来了什么好处?就拿大家比较关注的单细胞RNA-seq来说:
1.在传统的研究方法中,我们经常根据标记基因和细胞形态来区分不同的细胞类型,但这种方法无疑是有争议的。单细胞测序技术可以更准确、无偏倚地对细胞进行聚类。特别是免疫学、肿瘤学、遗传学方面的研究,会带来很大的影响。
2.分析稀有细胞,尤其是特定时间空环境下的细胞。如从环境中取样的微生物。
3.临床上,体外受精胚胎在植入前进行筛选。
4.基于循环肿瘤细胞诊断癌症,大力推广新型循环肿瘤细胞检测技术。尤其是治疗前后循环肿瘤细胞类型和数量的变化具有重要的预后价值。
5.大规模测序是通过传统的测序方法进行的。希望利用这些数据冲击重量级期刊的小伙伴们注意,单细胞测序已经成为高分期刊出版的井喷,技术在几年后会更加成熟。
2.要实现单细胞转录组测序,需要解决两个难题:
1.PCR偏倚:单个细胞含有约10pg的总RNA,80%以上的信息是rRNA。从单细胞RNA到文库,意味着核酸的扩增量要达到一百万倍以上。然而,在这种高扩增水平下,不引入聚合酶链反应偏差始终是一个大问题。
我们可以考虑一下。如果两个样本的基因表达相同,但A和B的扩增效率分别为99%和97%,经过30个循环的扩增,两者相差1.84倍。我们在分析差异基因的时候,如果选择1.5倍作为差异基因的标准,那么没有差异的基因就会有差异。
2.去除rRNA:rRNA占总RNA的80%以上。如果不加选择地进行反转录、再扩增和建立数据库,通过测序获得的大多数序列可能是rRNA序列。但是,一般来说,如果你更关心编码基因的序列,比如mRNA,rRNA序列并不能给我们带来有效的信息,可以说是没有用的。
分别介绍了以下三种单细胞转录组扩增技术:
智能放大技术:

SMART扩增技术的核心技术是设计两个专用引物。结合MMLV逆转录酶进行逆转录。
专用引物1由中间的polyte序列加上一个通用序列和3’端的两个简并碱基组成,但polyte 3’端倒数第二个碱基是A、C、G的简并碱基而不是T,倒数第二个碱基是简并碱基。这样做的好处是可以在mRNA的3’端和聚(A)的尾部的连接处结合,而在mRNA的其他地方不结合。这确保了逆转录的起始位置恰好是在mRNA 3’末端的序列终止位置。MMLV逆转录酶有一个特点,当它被转录到基因的5’端时,它会在新合成的基因的3’端增加几个碳碱基。
专用引物2由一个通用序列和其3’端的三个非脱氧G碱基组成,即RNA和RNA的G碱基,而不是DNA的G碱基。该引物可以与新合成的cDNA 3’端的几个碳碱基互补杂交,然后引导该MMLV酶再次进行聚合。复制的结果是获得双链cDNA。
这种双链cDNA的两端已经与我们设计的PCR引物序列相连,然后加入常规的PCR引物进行常规的PCR扩增和常规的PCR扩增,获得大量的DNA。然后,像传统的DNA数据库一样,它可以被超声波中断,在计算机上建立和测序。
SMART技术获得的主要信息是mRNA信息,大部分LncRNA信息都会丢失。SMART技术对RNA的质量要求很高,如果RNA被降解,mrna 5’端的信息就会丢失。通用引物技术可以保证扩增的均匀性,但不能分析PCR引入的突变。
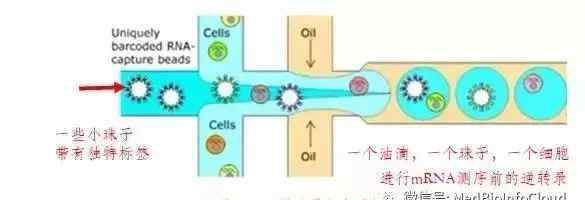
10倍基因组技术:
首先,将特定的脱氧核糖核酸片段接种在凝胶珠上,凝胶珠由三部分组成:条形码、UMI和多聚体。条形码长度为16个碱基。条形码有400万种,一个微珠对应一种条形码。通过这400万种条形码,可以区分凝胶微珠。UMI是一个随机序列,这意味着每个DNA分子都有自己的UMI序列。UMI有10个碱基长,有100万种序列变化。UMI的功能是区分哪些读数来自原始的基因分子,基因片段是重复的还是复制的,它们是真正的单核苷酸多态性位点还是聚合酶链反应产生的突变。单个细胞和单个凝胶珠用10×基因组学仪器通过油相混合在一起,形成油包水的小液滴,然后细胞膜破裂,释放细胞中的mRNA。游离的基因与水滴中的水相混合,也就是说,它与逆转录酶、结合在凝胶珠上的核酸引物和脱氧核糖核酸底物接触。
然后,发生反转录反应。mRNA与凝胶珠上标记的DNA分子结合,在逆转录酶的作用下反向转录。将这种乳液中的水相全部泵出,也就是将所有标记好的cDNA分子全部泵出,然后将这些cDNA分子加上接头,经PCR扩增,制成illumina测序文库,放在Illumina测序仪上测序。测序后,对数据进行分析。
10×基因组学技术可以一次性获得大量的大细胞数据,但只能获得mRNA信息,LncRNA的大部分信息丢失。UMI技术可以很好地去除被认为是由分析和聚合酶链反应中的重复引入的单核苷酸多态性位点。RNA的质量也高,降解也会造成5’-末端信息的丢失。
任何耗尽技术

Anydeplete技术首先使用随机引物进行单链合成。核苷酸类似物被引入到单链合成中用于酶消化,核苷酸类似物也被引入到双链合成中以确保链特异性。然后,在两端添加一个接头,接头的一条链还携带用于酶促降解的核苷酸类似物。形成单链文库时,设计特异性引物与rRNA形成的文库结合,经过一轮退火延伸,rRNA文库形成双链结构。反向接头上有特定的限制位点。当识别出限制性位点并切断接头时,由rRNA形成的文库不具有完整的接头,而其它文库具有完整的接头。通过聚合酶链反应扩增和富集,可以获得所需的信息。和10×基因组学一样,any delinet技术包含分子标签,可以分析复制和PCR产生的突变位点。
anydelete技术可用于可降解样品,确保5’端和3’端信息的完整性,同时获得mRNA和LncRNA信息。如果只需要基因信息,任何删除技术都会造成一些数据浪费。
参考:
1 . https://www . Sohu . com/a/332085879 _ 811044
1.《deplete 单细胞测序技术原理》援引自互联网,旨在传递更多网络信息知识,仅代表作者本人观点,与本网站无关,侵删请联系页脚下方联系方式。
2.《deplete 单细胞测序技术原理》仅供读者参考,本网站未对该内容进行证实,对其原创性、真实性、完整性、及时性不作任何保证。
3.文章转载时请保留本站内容来源地址,https://www.lu-xu.com/jiaoyu/731816.html